Bioinformática - Análise Filogenética com Clustalx
O Clustalx é um software para o alinhamento de sequências nucleotídicas, peptídicas e analises filogênicas. O Clustalx é software semi-livre segundo a sua forma de licenciamento. Embora possua características de software livre como permissão de execução de programa, não há permissão de uso para fins comerciais.
[ Hits: 27.906 ]
Por: José Cleydson Ferreira da Silva em 08/07/2010

Usando o Clustalx



- Gap Opening- penalização de escore para iniciar uma região de "Gap".
- Gap Extension- penalização de escore para estender uma região de "Gap" (normalmente menor que o Gap Opening)
- Delay Divergent Sequences - atraso de alinhamento de sequências divergentes que somente serão alinhadas após as outras sequências (porcentagem de identidade abaixo da qual a sequência será considerada divergente)
- Transition Weight (somente DNA) - da a transições (A<->G, T<->C) um escore diferente de 0.
- Use negative matrix - permite o uso de matrizes negativas, importante quando as sequências forem relacionadas somente em uma pequena porção. Em condições normais prejudica um pouco o alinhamento.
- Protein Weight Matrix - matriz de substituição a ser utilizada. Note que você só escolhe a serie de matriz: Blosum, PAM etc, o tipo de matriz dentro desta série (por exemplo Blosum 62, blosum 80) é escolhido automaticamente pelo programa.
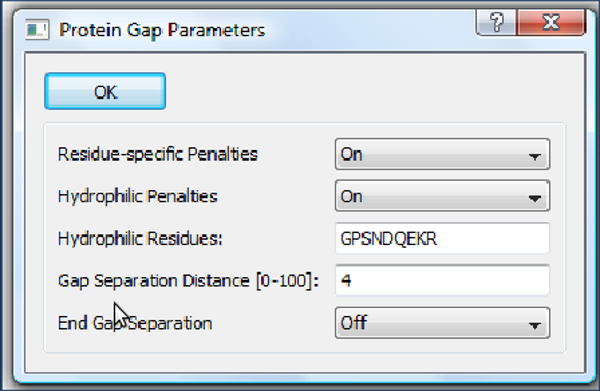
- Residue-specific Penalties - considera a vizinhança de alguns resíduos como mais ou menos favorável para a abertura de "Gaps".
- Hydrophilic Penalties - aumenta a chance de Gaps em regiões ricas em resíduos hidrofílicos que, usualmente, representam regiões menos estruturadas.
- Hydrophilic Residues - especifica quais resíduos são considerados hidrofílicos.
- Gap Separation Distance - número de resíduos de distância de uma região com Gap na qual é penalizada uma nova abertura de Gap.
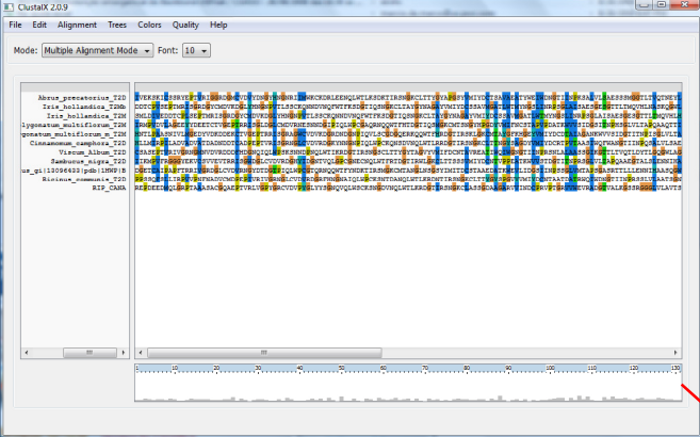
A apresentação de resultado de um alinhamento no ClustalX é representada por símbolos que indicam a conservação e os resíduos ou grupos de resíduos em uma coluna.
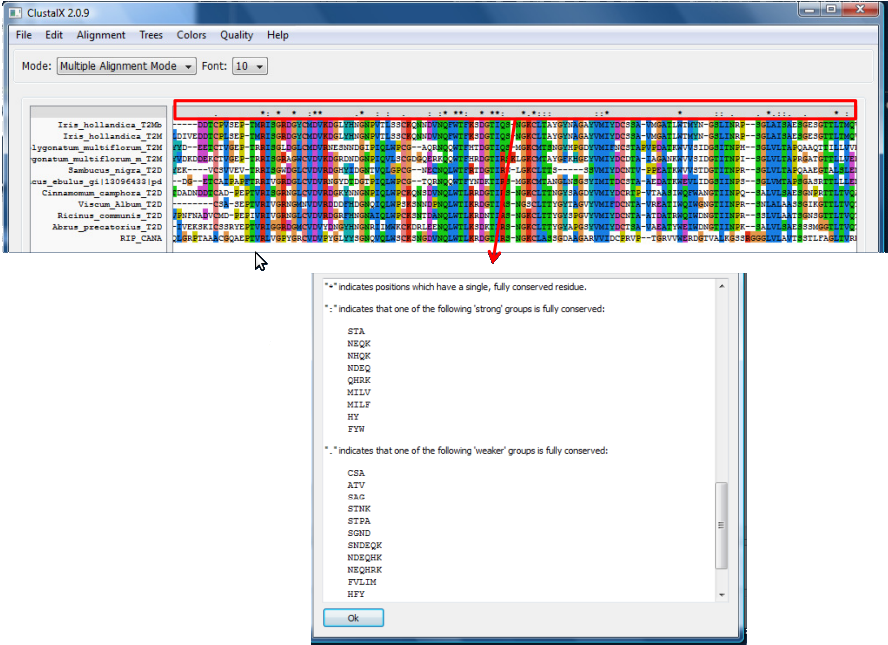
- Low Scoring Segments - mostra, em cinza, regiões com baixo escore e que, portanto, não seriam muito confiáveis
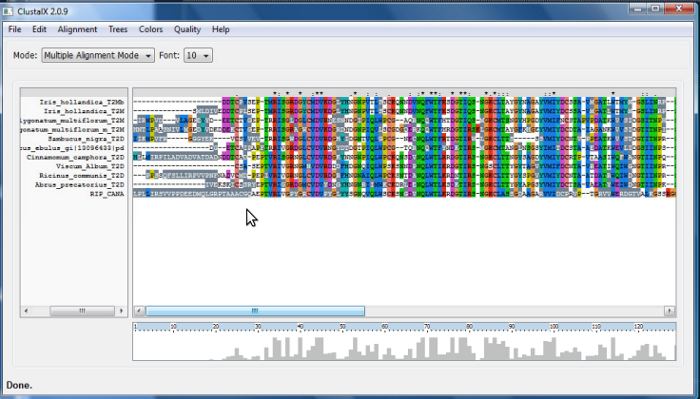
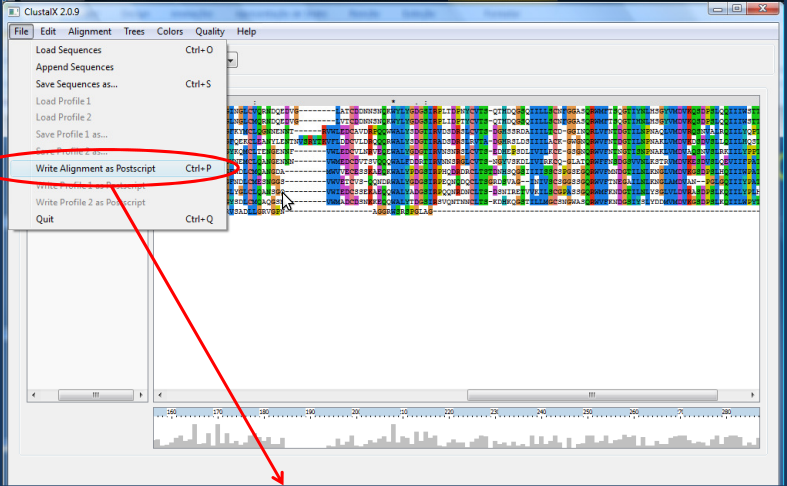
2. Usando o Clustalx
3. Sobre o autor
Bing: Medindo velocidade da conexão no Linux
Implementando servidor de aplicações PHP utilizando Zend Framework
Implementando rotas estáticas no Linux com route
Gerência de projetos com Redmine
Acessando desktop remotamente com noMachine
Terminais leves com LTSP - Linux Terminal Server Project
MiniDLNA - Servidor de mídia descomplicado
Nenhum comentário foi encontrado.
Patrocínio

Destaques
Artigos
Compartilhando a tela do Computador no Celular via Deskreen
Como Configurar um Túnel SSH Reverso para Acessar Sua Máquina Local a Partir de uma Máquina Remota
Configuração para desligamento automatizado de Computadores em um Ambiente Comercial
Dicas
Como renomear arquivos de letras maiúsculas para minúsculas
Imprimindo no formato livreto no Linux
Vim - incrementando números em substituição
Efeito "livro" em arquivos PDF
Como resolver o erro no CUPS: Unable to get list of printer drivers
Tópicos
Não to conseguindo resolver este problemas ao instalar o playonelinux (1)
Excluir banco de dados no xampp (1)
Top 10 do mês
-

Xerxes
1° lugar - 64.119 pts -

Fábio Berbert de Paula
2° lugar - 49.989 pts -

Buckminster
3° lugar - 17.299 pts -

Mauricio Ferrari
4° lugar - 15.004 pts -
Alberto Federman Neto.
5° lugar - 13.575 pts -

Diego Mendes Rodrigues
6° lugar - 13.367 pts -

Daniel Lara Souza
7° lugar - 12.759 pts -

Andre (pinduvoz)
8° lugar - 10.143 pts -

edps
9° lugar - 10.158 pts -

Alessandro de Oliveira Faria (A.K.A. CABELO)
10° lugar - 10.010 pts
Scripts
[Python] Automação de scan de vulnerabilidades
[Python] Script para analise de superficie de ataque
[Shell Script] Novo script para redimensionar, rotacionar, converter e espelhar arquivos de imagem
[Shell Script] Iniciador de DOOM (DSDA-DOOM, Doom Retro ou Woof!)
[Shell Script] Script para adicionar bordas às imagens de uma pasta
A maior comunidade GNU/Linux da América Latina! Artigos, dicas, tutoriais, fórum, scripts e muito mais. Ideal para quem busca auto-ajuda.
Site hospedado por: