Montagem de Cluster
Artigo referente à montagem completa de um Cluster com o sistema Debian Wheezy, podendo ser adaptado para outras distribuições.

Parte 7: Cluster 5
11. MPICH
MPI (Message Passing Interface), em uma tradução livre, Interface de Passagem de Mensagens. É uma biblioteca com funções para troca de mensagens que faz a comunicação e a sincronização de processos em um Cluster paralelo.
Essa é a biblioteca que fará todo o trabalho de processamento no Cluster. Essa é a biblioteca que transforma um conjunto de máquinas em um Cluster.
Os objetivos do MPICH é fornecer uma implementação MPI que suporte eficientemente, diferentes plataformas de computação e comunicação, incluindo grupos de commodities (sistemas de Desktop, sistemas de memória compartilhada e arquiteturas multicore), redes de alta velocidade e sistemas de computação High-end de propriedade (Blue Gene, Cray); e permitir a pesquisa de ponta em MPI através de uma estrutura modular fácil de estender para outras implementações derivadas.
Servidor e nós:
# cd /usr/src
# wget http://www.mpich.org/static/downloads/3.0.4/mpich-3.0.4.tar.gz
# tar -xzvf mpich-3.0.4.tar.gz
# ls
# cd mpich-3.0.4
#./configure --help | less
# ./configure --prefix=/opt/mpich --enable-shared --enable-f95 --enable-threads=runtime --enable-romio --enable-nemesis-shm-collectives --enable-debuginfo CC=gcc CXX=g++ FC=gfortran F77=gfortran --with-pvfs2=/opt/pvfs2 --with-file-system="pvfs2+nfs" --with-thread-package=posix --with-device=ch3:nemesis --with-java=/etc/java-7-openjdk # Somente se for trabalhar com Java
Se não tiver erros, terminará com:
# make
O make deverá terminar com a frase abaixo sem nenhuma mensagem de erro antes dela:
# make install
O make install, idem ao make, não deverá apresentar erros.
Configurando as variáveis:
# vim ~/.bashrc
E acrescentar, também, o caminho na variável LD_LIBRARY_PATH:
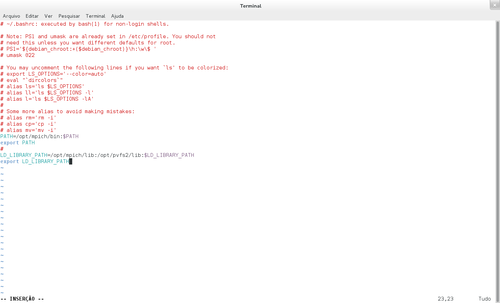
O arquivo bashrc acima com as variáveis, é o mesmo para o servidor e os nós.
Reinicie:
# shutdown -r now
Vendo as informações:
# mpiexec -info
Vamos testar nossa instalação, compilando e executando:
# cd /usr/src/mpich-3.0.4/examples
# ls
# mpicc -o cpi cpi.c # Esse arquivo deve ser compilado em todas as máquinas; acesse uma por uma ou utilize o seguinte comando:
# mpicc -o -hostfile /opt/hostfile /usr/src/mpich-3.0.4/examples/cpi.c cpi
O arquivo cpi.c realiza cálculo do PI.
Testando:
# mpirun -hostfile /opt/hostfile -n 7 /usr/src/mpich-3.0.4/examples/cpi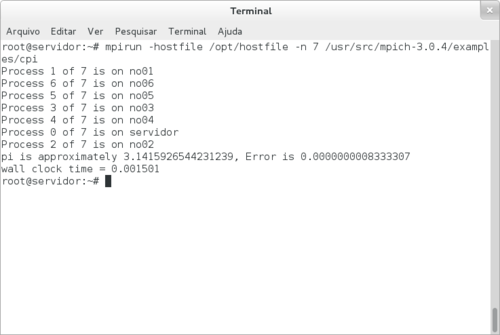
Para ver o manual, execute:
# man mpicc
Ou:
# man mpi_Barrier
Ou:
# man mpiexec
Para remover, em caso de erros e começar de novo:
# cd /usr/src/mpich-3.0.4
# make uninstall
# make distclean
# cd /usr/src/pvfs-2.8.2
# make distclean
E remova os seguintes arquivos com rm -rf:
Reinicie a máquina e recomece do princípio, as duas instalações criando novamente os diretórios /opt/mpich e /opt/pvfs2.
12. Ganglia
O Ganglia é um sistema de monitoramento distribuído escalável para sistemas de computação de alto desempenho, como Clusters e grids. Ele é baseado em um projeto hierárquico dirigido a Clusters. Ele aproveita as tecnologias amplamente utilizadas, tais como XML para representação de dados, XDR para compacto, transporte de dados portátil e RRDtool para o armazenamento e visualização de dados.
Ele usa as estruturas de dados e algoritmos projetados cuidadosamente para alcançar baixos custos gerais por nós e alta concorrência. A implementação é robusta e foi portada para um amplo conjunto de sistemas operacionais e arquiteturas de processadores, e está atualmente em uso em milhares de grupos em todo o mundo.
Ele tem sido usado para ligar os Clusters através de campi universitários e em todo o mundo, e pode ser escalado para lidar com grupos com 2000 nós.
Nos nós:
# apt-get update
# apt-get install ganglia-monitor
No servidor:
# apt-get update
# apt-get install apache2
Testando o Apache: abra o navegador e digite: localhost, deverá aparecer: It works.
# apt-get install php5 libapache2-mod-php5
Crie o arquivo:
# vim /var/www/info.php
Coloque dentro dele:
Salve e saia. Reinicie o Apache:
# /etc/init.d/apache2 restart
Abra o navegador e digite localhost/info.php, deverá aparecer a página de informações do PHP.
Instalando o Ganglia:
# apt-get install ganglia-webfrontend ganglia-monitor
Irá pedir duas vezes para reiniciar o Apache 2, faça.
Vamos copiar o arquivo necessário:
# cp /etc/ganglia-webfrontend/apache.conf /etc/apache2/sites-enabled/ganglia.conf
Alterar o arquivo /etc/ganglia/gmod.conf no servidor e copiar para os nós. É extremamente necessário, tendo em vista que a conexão remota fica muito lenta, devido ao IP 239.2.11.71.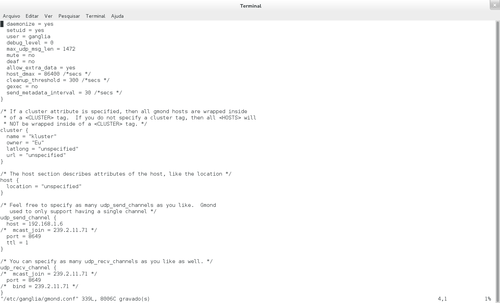
A parte que nos interessa alterar, está na imagem acima. Comente as linhas onde tem o IP 239.2.11.71, ou similar, e configure o host com o IP do servidor.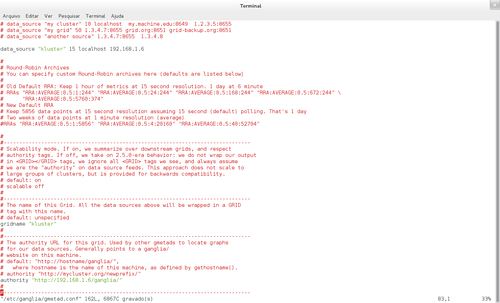
Altere o arquivo /etc/ganglia/gmetad.conf no servidor, deixando como na imagem acima, não sendo necessário copiar para os nós.
Para reiniciar o serviço nos nós:
# service ganglia-monitor restart
Reiniciar o Apache e o Ganglia no servidor:
# /etc/init.d/apache2 restart
# service gmetad restart
A partir daí é só digitar no navegador localhost/ganglia.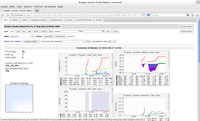
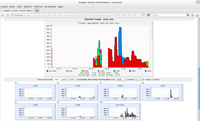
MPI (Message Passing Interface), em uma tradução livre, Interface de Passagem de Mensagens. É uma biblioteca com funções para troca de mensagens que faz a comunicação e a sincronização de processos em um Cluster paralelo.
Essa é a biblioteca que fará todo o trabalho de processamento no Cluster. Essa é a biblioteca que transforma um conjunto de máquinas em um Cluster.
Os objetivos do MPICH é fornecer uma implementação MPI que suporte eficientemente, diferentes plataformas de computação e comunicação, incluindo grupos de commodities (sistemas de Desktop, sistemas de memória compartilhada e arquiteturas multicore), redes de alta velocidade e sistemas de computação High-end de propriedade (Blue Gene, Cray); e permitir a pesquisa de ponta em MPI através de uma estrutura modular fácil de estender para outras implementações derivadas.
Servidor e nós:
# cd /usr/src
# wget http://www.mpich.org/static/downloads/3.0.4/mpich-3.0.4.tar.gz
# tar -xzvf mpich-3.0.4.tar.gz
# ls
# cd mpich-3.0.4
#./configure --help | less
# ./configure --prefix=/opt/mpich --enable-shared --enable-f95 --enable-threads=runtime --enable-romio --enable-nemesis-shm-collectives --enable-debuginfo CC=gcc CXX=g++ FC=gfortran F77=gfortran --with-pvfs2=/opt/pvfs2 --with-file-system="pvfs2+nfs" --with-thread-package=posix --with-device=ch3:nemesis --with-java=/etc/java-7-openjdk # Somente se for trabalhar com Java
Se não tiver erros, terminará com:
Configuration completed
# make
O make deverá terminar com a frase abaixo sem nenhuma mensagem de erro antes dela:
make[1]: Saindo do diretório '/usr/src/mpich-3.0.4'
# make install
O make install, idem ao make, não deverá apresentar erros.
Configurando as variáveis:
# vim ~/.bashrc
PATH=/opt/mpich/bin:$PATH
export PATH
export PATH
E acrescentar, também, o caminho na variável LD_LIBRARY_PATH:
/opt/mpich/lib

O arquivo bashrc acima com as variáveis, é o mesmo para o servidor e os nós.
Reinicie:
# shutdown -r now
Vendo as informações:
# mpiexec -info
Vamos testar nossa instalação, compilando e executando:
# cd /usr/src/mpich-3.0.4/examples
# ls
# mpicc -o cpi cpi.c # Esse arquivo deve ser compilado em todas as máquinas; acesse uma por uma ou utilize o seguinte comando:
# mpicc -o -hostfile /opt/hostfile /usr/src/mpich-3.0.4/examples/cpi.c cpi
O arquivo cpi.c realiza cálculo do PI.
Testando:
# mpirun -hostfile /opt/hostfile -n 7 /usr/src/mpich-3.0.4/examples/cpi
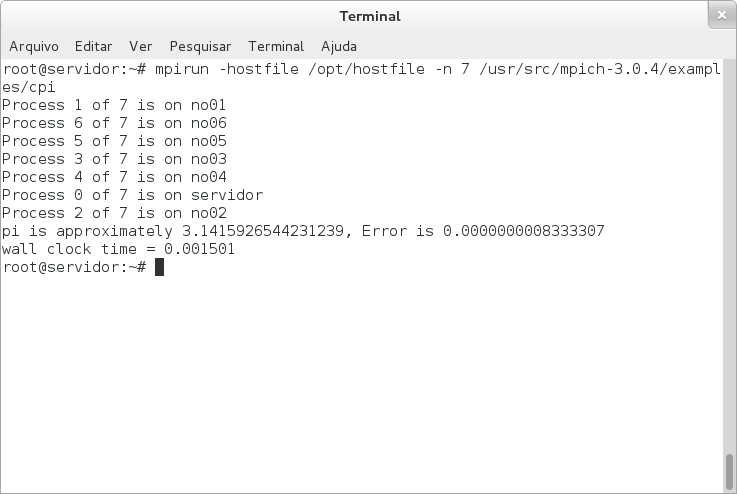
Para ver o manual, execute:
# man mpicc
Ou:
# man mpi_Barrier
Ou:
# man mpiexec
Para remover, em caso de erros e começar de novo:
# cd /usr/src/mpich-3.0.4
# make uninstall
# make distclean
# cd /usr/src/pvfs-2.8.2
# make distclean
E remova os seguintes arquivos com rm -rf:
- /usr/src/mpich-3.0.4
- /usr/src/pvfs-2.8.2
- /opt/mpich
- /opt/pvfs2
- /usr/local/sbin/pvfs2-server
Reinicie a máquina e recomece do princípio, as duas instalações criando novamente os diretórios /opt/mpich e /opt/pvfs2.
12. Ganglia
O Ganglia é um sistema de monitoramento distribuído escalável para sistemas de computação de alto desempenho, como Clusters e grids. Ele é baseado em um projeto hierárquico dirigido a Clusters. Ele aproveita as tecnologias amplamente utilizadas, tais como XML para representação de dados, XDR para compacto, transporte de dados portátil e RRDtool para o armazenamento e visualização de dados.
Ele usa as estruturas de dados e algoritmos projetados cuidadosamente para alcançar baixos custos gerais por nós e alta concorrência. A implementação é robusta e foi portada para um amplo conjunto de sistemas operacionais e arquiteturas de processadores, e está atualmente em uso em milhares de grupos em todo o mundo.
Ele tem sido usado para ligar os Clusters através de campi universitários e em todo o mundo, e pode ser escalado para lidar com grupos com 2000 nós.
Nos nós:
# apt-get update
# apt-get install ganglia-monitor
No servidor:
# apt-get update
# apt-get install apache2
Testando o Apache: abra o navegador e digite: localhost, deverá aparecer: It works.
# apt-get install php5 libapache2-mod-php5
Crie o arquivo:
# vim /var/www/info.php
Coloque dentro dele:
<?php
phpinfo();
?>
phpinfo();
?>
Salve e saia. Reinicie o Apache:
# /etc/init.d/apache2 restart
Abra o navegador e digite localhost/info.php, deverá aparecer a página de informações do PHP.
Instalando o Ganglia:
# apt-get install ganglia-webfrontend ganglia-monitor
Irá pedir duas vezes para reiniciar o Apache 2, faça.
Vamos copiar o arquivo necessário:
# cp /etc/ganglia-webfrontend/apache.conf /etc/apache2/sites-enabled/ganglia.conf
Alterar o arquivo /etc/ganglia/gmod.conf no servidor e copiar para os nós. É extremamente necessário, tendo em vista que a conexão remota fica muito lenta, devido ao IP 239.2.11.71.

A parte que nos interessa alterar, está na imagem acima. Comente as linhas onde tem o IP 239.2.11.71, ou similar, e configure o host com o IP do servidor.

Altere o arquivo /etc/ganglia/gmetad.conf no servidor, deixando como na imagem acima, não sendo necessário copiar para os nós.
Para reiniciar o serviço nos nós:
# service ganglia-monitor restart
Reiniciar o Apache e o Ganglia no servidor:
# /etc/init.d/apache2 restart
# service gmetad restart
A partir daí é só digitar no navegador localhost/ganglia.

